Every cell holds a unique story written in its RNA. RNA sequencing (RNA-Seq) reveals gene expression in unprecedented detail, helping researchers understand disease, identify biomarkers, and develop new therapies. To turn complex data into meaningful discoveries, Source Genomics leverages Next Generation Sequencing (NGS) to deliver high-quality RNA sequencing with expert bioinformatics, advanced technology, and flexible solutions tailored to your research.
RNA-Seq Services
We have a comprehensive suite of RNA-Seq solutions tailored to diverse research applications:
Total RNA Sequencing
Captures both coding and non-coding RNA, offering a complete transcriptomic profile.
Transcriptome Sequencing
Focuses on mRNA, enabling gene expression analysis and the discovery of novel transcripts.
Small RNA Sequencing
Identifies microRNAs and other small regulatory RNAs essential for gene regulation.
Single Cell RNA Sequencing
Provides a detailed view of gene expression at the individual cell level, revealing cellular diversity.
Low Input RNA Sequencing
Enables sequencing from minimal RNA quantities, ensuring accurate results even from limited samples.
Our RNA sequencing services generate high-quality, reproducible data tailored to the demands of oncology, neuroscience, immunology, developmental biology, and beyond. With flexible sequencing depths and customisable analysis options, we ensure precision and adaptability for diverse scientific inquiries.
Contact us to discuss your project today. Our team is committed to the highest quality standards and fast turnaround times, ensuring your research needs are always our top priority.
How Our RNA Sequencing Analysis Works
Here at Source Genomics, a streamlined sequencing workflow ensures data integrity at every step:
- RNA Quality Control – Rigorous assessment of RNA integrity and purity to confirm sample suitability.
- Library Preparation - High-efficiency library construction using 2 picograms of complete RNA
- Sequencing – High-depth sequencing with 150bp paired-end reads and 30 million clusters per sample.
- Data Processing & Bioinformatics – Demultiplexing, quality filtering, and advanced bioinformatics options for deeper insights.
Every sequencing project is customised to match the specific requirements of your research. From raw sequencing reads to full bioinformatics analysis, we provide the level of detail and support needed to advance your work.
Benefits
Partnering with Source Genomics for RNA sequencing gives you:
Unmatched Data Quality
ISO-accredited labs and rigorous QC processes ensure reliable, high-resolution sequencing results.
Flexible & Scalable Solutions
Tailorable sequencing parameters, read depths, and analysis options to suit your research.
Quick & Efficient Turnaround
Optimised workflows ensure rapid processing times to keep your projects moving forward.
Expert Scientific Guidance
Dedicated specialists provide support at every stage, including experimental design, sequencing execution, and data interpretation.
Overview
Sequencing package details
At Source Genomics we realise that the standard package does not always fit the customer needs – we offer the flexibility to increase/decrease both the number of reads and read length required. Our standard package includes:
• Initial QC assessment of RNA
• Library preparation from as little as 2pg total RNA
• Sequencing to a depth of 30 million clusters with PE150bp reads
• Demultiplex and return of data
RNA extraction from various sample sources as well as advanced Bio-informatic analysis is also available to support your entire project needs.
The process
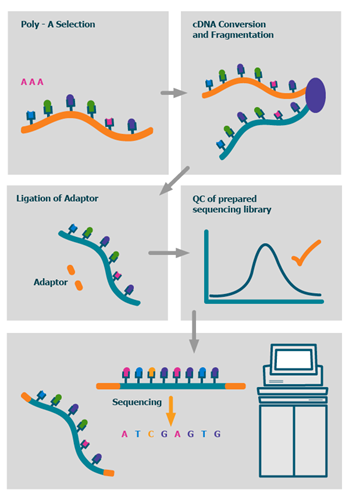
We also offer advanced BioIT variant analysis packages as well as fully bespoke BioIT solutions.
BioInformatics
Digital gene expression
Quantification of transcripts in a sample using RNA-Seq data
This analysis uses RNA-Seq data to quantify expression levels by comparison to a reference genome, or de novo transcriptome assembly if a reference genome is not available. Raw reads are trimmed, aligned and quantified by gene-wise read counting using the featureCounts model. You will receive raw (fastq) and aligned (bam) files for each sample as well as text files detailing transcripts per million (TPM) or fragments per kb per million mapped reads (FPKM). A bioinformatics report will provide further detail about the tools used, alignment metrics and a summary of results.
Differential gene expression
Pairwise comparison of gene expression between sample groups
This analysis is similar to digital gene expression, but includes a comparative analysis between groups of samples. You will receive raw (fastq), aligned (bam) files and text files of gene counts as well as a PCA plot of all samples, a sample-to-sample distance heatmap and MA plots and differential expression heatmaps for each comparison. A bioinformatics report will provide further detail about the tools used, alignment metrics and a summary of results.
Other gene expression methods
Bespoke analyses of other RNA-Seq data, such as (differential) miRNA or methylation quantification; identification of splice variants; detection and quantification of fusion genes.
Transcriptome assembly
De novo or reference-guided assembly of RNAseq data into one transcriptome
Depending on the availability of a reference sequence, reads are de novo assembled, or against a reference genome (or a combination of the two) to create sequence scaffolds.
The analysis includes quality trimming of raw reads, assembly into contigs – mapping against a reference, if available – filtering and creating sequence scaffolds. You will receive raw (fastq) files, the assembled sequence file (fasta) and a bioinformatics report detailing the tools used and contig size statistics.
How to order
Contact us today and one of our skilled account managers will be in touch with a free consultation including further information and pricing details.
Payment options:
Payment can be made by credit card or purchase order number.
FAQ's:
View our frequently asked questions for more information.
Sample requirements:
We ask optimally for at least 300ng of total RNA at 6 ng/μl concentration in a minimum volume of 50 μl volume with a RIN of 7 or above. Please contact if your samples do not reach these requirements to discuss alternative approaches. Higher concentrations can be submitted and will be diluted accordingly. For full details click here.
Discover the Power of RNA Sequencing
Unlock the full potential of RNA sequencing with Source Genomics. Contact our team today to discuss your project, explore customised sequencing options, and get a tailored quote. Let’s advance your research together!
Contact us today and one of our skilled account managers will be in touch with a free consultation including further information and pricing details.
Next Generation Sequencing
Source BioScience is one of Europe’s leading providers of commercial sequencing, offering Next Generation Sequencing services from our ISO accredited laboratories. We offer NGS services on the most prominent platforms including Illumina’s NovaSeq, NextSeq and MiSeq.